

Search
- Page Path
-
- HOME
- Search
- Case Report
- Genetics and Metabolism
- A compound heterozygous mutation in the
FMO3 gene: the first pediatric case causes fish odor syndrome in Korea - Ji Hyun Kim, Sung Min Cho, Jong-Hee Chae
- Clin Exp Pediatr. 2017;60(3):94-97. Published online March 27, 2017
-
Trimethylaminuria (TMAuria), known as “fish odor syndrome,” is a congenital metabolic disorder characterized by an odor resembling that of rotting fish. This odor is caused by the secretion of trimethylamine (TMA) in the breath, sweat, and body secretions and the excretion of TMA along with urine. TMAuria is an autosomal recessive disorder caused by mutations in flavin-containing monooxygenase 3 (
FMO3 )....
- Review Article
- Nephrology (Genitourinary)
- Genetics of hereditary nephrotic syndrome: a clinical review
- Tae-Sun Ha
- Clin Exp Pediatr. 2017;60(3):55-63. Published online March 27, 2017
-
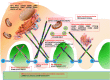
Advances in podocytology and genetic techniques have expanded our understanding of the pathogenesis of hereditary steroid-resistant nephrotic syndrome (SRNS). In the past 20 years, over 45 genetic mutations have been identified in patients with hereditary SRNS. Genetic mutations on structural and functional molecules in podocytes can lead to serious injury in the podocytes themselves and in adjacent structures, causing sclerotic...
- Case Report
- Neurology
- Concurrency of Guillain-Barre syndrome and acute transverse myelitis: a case report and review of literature
- Orkun Tolunay, Tamer Çelik, Ümit Çelik, Mustafa Kömür, Zeynep Tanyeli, Abdurrahman Sönmezler
- Clin Exp Pediatr. 2016;59(Suppl 1):S161-S164. Published online November 30, 2016
-
Guillain-Barré syndrome and acute transverse myelitis manifest as demyelinating diseases of the peripheral and central nervous system. Concurrency of these two disorders is rarely documented in literature. A 4-year-old girl presenting with cough, fever, and an impaired walking ability was admitted to hospital. She had no previous complaints in her medical history. A physical examination revealed lack of muscle strength...
- Nephrology (Genitourinary)
- Posterior reversible encephalopathy syndrome caused by presumed Takayasu arteritis
- Ki Wuk Lee, Sang Taek Lee, Heeyeon Cho
- Clin Exp Pediatr. 2016;59(Suppl 1):S145-S148. Published online November 30, 2016
-
Takayasu arteritis (TA) is a chronic inflammatory disease of unknown etiology that affects mainly the aorta, main aortic branches, and pulmonary arteries. Diverse neurological manifestations of TA have rarely been reported in children. Posterior reversible encephalopathy syndrome (PRES) is a neuroradiological condition that presents with headache, seizure, visual disturbances, and characteristic lesions on imaging. Inflammatory condition and severe hypertension in...
- Neurology
- A pediatric case of idiopathic Harlequin syndrome
- Ju Young Kim, Moon Souk Lee, Seung Yeon Kim, Hyun Jung Kim, Soo Jin Lee, Chur Woo You, Jon Soo Kim, Ju Hyung Kang
- Clin Exp Pediatr. 2016;59(Suppl 1):S125-S128. Published online November 30, 2016
-
Harlequin syndrome, which is a rare disorder caused by dysfunction of the autonomic system, manifests as asymmetric facial flushing and sweating in response to heat, exercise, or emotional factors. The syndrome may be primary (idiopathic) with a benign course, or can occur secondary to structural abnormalities or iatrogenic factors. The precise mechanism underlying idiopathic harlequin syndrome remains unclear. Here, we...
- Endocrinology
- Concomitant occurrence of Turner syndrome and growth hormone deficiency
- Jung Yu, Ha Young Shin, Chong Guk Lee, Jae Hyun Kim
- Clin Exp Pediatr. 2016;59(Suppl 1):S121-S124. Published online November 30, 2016
-
Turner syndrome (TS) is a genetic disorder in phenotypic females that has characteristic physical features and presents as partial or complete absence of the second sex chromosome. Growth hormone deficiency (GHD) is a condition caused by insufficient release of growth hormone from the pituitary gland. The concomitant occurrence of TS and GHD is rare and has not yet been reported...
- Hashimoto thyroiditis with an unusual presentation of cardiac tamponade in Noonan syndrome
- Mi Ji Lee, Byung Young Kim, Jae Sook Ma, Young Earl Choi, Young Ok Kim, Hwa Jin Cho, Chan Jong Kim
- Clin Exp Pediatr. 2016;59(Suppl 1):S112-S115. Published online November 30, 2016
-
Noonan syndrome is an autosomal dominant, multisystem disorder. Autoimmune thyroiditis with hypothyroidism is an infrequent feature in patients with Noonan syndrome. A 16-year-old boy was admitted because of chest discomfort and dyspnea; an echocardiogram revealed pericardial effusion. Additional investigations led to a diagnosis of severe hypothyroidism due to Hashimoto thyroiditis. The patient was treated with L-thyroxine at 0.15 mg daily....
- Nephrology (Genitourinary)
- A novel mutation of
CLCNKB in a Korean patient of mixed phenotype of Bartter-Gitelman syndrome - Hee-Won Cho, Sang Taek Lee, Heeyeon Cho, Hae Il Cheong
- Clin Exp Pediatr. 2016;59(Suppl 1):S103-S106. Published online November 30, 2016
-
Bartter syndrome (BS) is an inherited renal tubular disorder characterized by low or normal blood pressure, hypokalemic metabolic alkalosis, and hyperreninemic hyperaldosteronism. Type III BS is caused by loss-of-function mutations in
CLCNKB encoding basolateral ClC-Kb. The clinical phenotype of patients withCLCNKB mutations has been known to be highly variable, and cases that are difficult to categorize as type III...
- Eosinophilic gastroenteritis in an 18-year-old male with prolonged nephrotic syndrome
- Da Min Choi, Jung Eun Pyun, Hyung Eun Yim, Kee Hwan Yoo, Jung Ok Shim, Eun Jung Lee, Nam Hee Won
- Clin Exp Pediatr. 2016;59(Suppl 1):S72-S75. Published online November 30, 2016
-
Eosinophilic gastroenteritis is a rare disease characterized by prominent eosinophilic tissue infiltration of the gastrointestinal tract. Here, we report a case of eosinophilic gastroenteritis in an 18-year-old patient with prolonged nephrotic syndrome who presented with abdominal pain and peripheral hypereosinophilia. During the previous 2 years, he had visited local Emergency Department several times because of epigastric pain and nausea. He...
- Neurology
- A neonate with Joubert syndrome presenting with symptoms of Horner syndrome
- Narae Lee, Sang-Ook Nam, Young Mi Kim, Yun-Jin Lee
- Clin Exp Pediatr. 2016;59(Suppl 1):S32-S36. Published online November 30, 2016
-
Joubert syndrome (JS) is characterized by the “molar tooth sign” (MTS) with cerebellar vermis agenesis, episodic hyperpnea, abnormal eye movements, and hypotonia. Ocular and oculomotor abnormalities have been observed; however, Horner syndrome (HS) has not been documented in children with JS. We present the case of a 2-month-old boy having ocular abnormalities with bilateral nystagmus, left-dominant bilateral ptosis, and unilateral...
- Glucose transport 1 deficiency presenting as infantile spasms with a mutation identified in exon 9 of
SLC2A1 - Hyun Hee Lee, Yun Jung Hur
- Clin Exp Pediatr. 2016;59(Suppl 1):S29-S31. Published online November 30, 2016
-
Glucose transport 1 (GLUT-1) deficiency is a rare syndrome caused by mutations in the glucose transporter 1 gene (
SLC2A1 ) and is characterized by early-onset intractable epilepsy, delayed development, and movement disorder.De novo mutations and several hot spots in N34, G91, R126, R153, and R333 of exons 2, 3, 4, and 8 ofSLC2A1 are associated with this condition. Seizures,...
- Endocrinology
- Phelan-McDermid syndrome presenting with developmental delays and facial dysmorphisms
- Yoon-Myung Kim, In-Hee Choi, Jun Suk Kim, Ja Hye Kim, Ja Hyang Cho, Beom Hee Lee, Gu-Hwan Kim, Jin-Ho Choi, Eul-Ju Seo, Han-Wook Yoo
- Clin Exp Pediatr. 2016;59(Suppl 1):S25-S28. Published online November 30, 2016
-
Phelan-McDermid syndrome is a rare genetic disorder caused by the terminal or interstitial deletion of the chromosome 22q13.3. Patients with this syndrome usually have global developmental delay, hypotonia, and speech delays. Several putative genes such as the
SHANK3 ,RAB ,RABL2B , andIB2 are responsible for the neurological features. This study describes the clinical features and outcomes of Korean patients with...
- Neurology
- 1p36 deletion syndrome confirmed by fluorescence
in situ hybridization and array-comparative genomic hybridization analysis - Dong Soo Kang, Eunsim Shin, Jeesuk Yu
- Clin Exp Pediatr. 2016;59(Suppl 1):S14-S18. Published online November 30, 2016
-
Pediatric epilepsy can be caused by various conditions, including specific syndromes. 1p36 deletion syndrome is reported in 1 in 5,000–10,000 newborns, and its characteristic clinical features include developmental delay, mental retardation, hypotonia, congenital heart defects, seizure, and facial dysmorphism. However, detection of the terminal deletion in chromosome 1p by conventional G-banded karyotyping is difficult. Here we present a case of...
- Chromosome 11q13 deletion syndrome
- Yu-Seon Kim, Gun-Ha Kim, Jung Hye Byeon, So-Hee Eun, Baik-Lin Eun
- Clin Exp Pediatr. 2016;59(Suppl 1):S10-S13. Published online November 30, 2016
-
Chromosome 11q13 deletion syndrome has been previously reported as either otodental syndrome or oculo-oto-dental syndrome. The otodental syndrome is characterized by dental abnormalities and high-frequency sensorineural hearing loss, and by ocular coloboma in some cases. The underlying genetic defect causing otodental syndrome is a hemizygous microdeletion involving the
FGF3 gene on chromosome 11q13.3. Recently, a new form of severe deafness,...
- Immunology
- A familial case of Blau syndrome caused by a novel
NOD2 genetic mutation - Woojoong Kim, Eujin Park, Yo Han Ahn, Jiwon M. Lee, Hee Gyung Kang, Byung Joo Kim, Il-Soo Ha, Hae Il Cheong
- Clin Exp Pediatr. 2016;59(Suppl 1):S5-S9. Published online November 30, 2016
-
Blau syndrome (BS) is a rare autosomal dominant, inflammatory syndrome that is characterized by the clinical triad of granulomatous dermatitis, symmetric arthritis, and recurrent uveitis. Mutations in the nucleotide oligomerization domain 2 (
NOD2 ) gene are responsible for causing BS. To date, up to 30 Blau-associated genetic mutations have been identified within this gene. We report a novelNOD2 genetic mutation...
- Genetics and Metabolism
- A nonsense
PAX6 mutation in a family with congenital aniridia - Kyoung Hee Han, Hye Jin Lee, Il-Soo Ha, Hee Gyung Kang, Hae Il Cheong
- Clin Exp Pediatr. 2016;59(Suppl 1):S1-S4. Published online November 30, 2016
-
Congenital aniridia is a rare ocular malformation that presents with severe hypoplasia of the iris and various ocular manifestations. Most cases of congenital aniridia are known to be related to mutations in the paired box gene-6 (
PAX6 ), which is an essential gene in eye development. Herein, we report a familial case of autosomal dominant congenital aniridia with four affected members...
- Edentulous child with Allgrove syndrome: a rare case report
- Mohammad Vahedi, Shima Fathi, Hanif Allahbakhshi
- Clin Exp Pediatr. 2016;59(11):456-459. Published online November 18, 2016
-
Triple-A syndrome, also known as Allgrove syndrome, is a rare autosomal recessive disorder. The 3 features of this syndrome are achalasia, adrenal insufficiency, and alacrima. Achalasia could be the first manifestation of the triple-A syndrome; however, its etiology is unclear. Alacrima is generally asymptomatic but can be detected by obtaining patient history. Although adrenal insufficiency could have manifestations such as...
- Rheumatology
- Recurrent macrophage activation syndrome since toddler age in an adolescent boy with HLA B27 positive juvenile ankylosing spondylitis
- Joon Hyeong Park, Yu Mi Seo, Seung Beom Han, Ki Hwan Kim, Jung Woo Rhim, Nack Gyun Chung, Myung Shin Kim, Jin Han Kang, Dae Chul Jeong
- Clin Exp Pediatr. 2016;59(10):421-424. Published online October 17, 2016
-
Recurrent macrophage activation syndrome (MAS) is very rare. We present the case of an adolescent boy with human leukocyte antigen (HLA) B27-positive ankylosing spondylitis (AS), who experienced episodes of recurrent MAS since he was a toddler. A 16-year-old boy was admitted because of remittent fever with pancytopenia and splenomegaly after surgical intervention for an intractable perianal abscess. He had been...
- Original Article
- Cardiology
- Prediction of unresponsiveness to second intravenous immunoglobulin treatment in patients with Kawasaki disease refractory to initial treatment
- Euri Seo, Jeong Jin Yu, Hyun Ok Jun, Eun Jung Shin, Jae Suk Baek, Young-Hwue Kim, Jae-Kon Ko
- Clin Exp Pediatr. 2016;59(10):408-413. Published online October 17, 2016
-
Purpose This study investigated predictors of unresponsiveness to second-line intravenous immunoglobulin (IVIG) treatment for Kawasaki disease (KD).
Methods This was a single-center analysis of the medical records of 588 patients with KD who had been admitted to Asan Medical Center between 2006 and 2014. Related clinical and laboratory data were analyzed by univariate and multivariate logistic regression analyses.
Results Eighty (13.6%) of the 588 patients...
- Case Report
- Neurology
- Painful legs and moving toes syndrome in a 16-year-old girl
- Seung Soo Kim, Yong Seung Hwang, Young Chang Kim
- Clin Exp Pediatr. 2016;59(9):381-383. Published online September 21, 2016
-
Painful legs and moving toes (PLMT) syndrome is characterized by spontaneous movements of the digits and pain in one or both lower extremities. Of the reported cases, a majority of the patients was female, and the mean age of onset was 58 years. Only one pediatric case has been reported so far. Herein, we report the first adolescent case of...
- Original Article
- Cardiology
- Clinical outcome of patients with refractory Kawasaki disease based on treatment modalities
- Hyun Jung Kim, Hyo Eun Lee, Jae Won Yu, Hong Ryang Kil
- Clin Exp Pediatr. 2016;59(8):328-334. Published online August 24, 2016
-
Purpose Although a significant number of reports on new therapeutic options for refractory Kawasaki disease (KD) such as steroid, infliximab, or repeated intravenous immunoglobulin (IVIG) are available, their effectiveness in reducing the prevalence of coronary artery lesions (CAL) remains controversial. This study aimed to define the clinical characteristics of patients with refractory KD and to assess the effects of adjuvant therapy...
- Age-adjusted plasma N-terminal pro-brain natriuretic peptide level in Kawasaki disease
- Heul Jun, Kyung Ok Ko, Jae Woo Lim, Jung Min Yoon, Gyung Min Lee, Eun Jung Cheon
- Clin Exp Pediatr. 2016;59(7):298-302. Published online July 31, 2016
-
Purpose Recent reports showed that plasma N-terminal pro-brain natriuretic peptide (NT-proBNP) could be a useful biomarker of intravenous immunoglobulin (IVIG) unresponsiveness and coronary artery lesion (CAL) development in Kawasaki disease (KD). The levels of these peptides are critically influenced by age; hence, the normal range and upper limits for infants and children are different. We performed an age-adjusted analysis of plasma...
- Case Report
- Genetics and Metabolism
- A rare case of Sjogren-Larsson syndrome with recurrent pneumonia and asthma
- Azita Tavasoli, Shirin Sayyahfar, Babak Behnam
- Clin Exp Pediatr. 2016;59(6):276-279. Published online June 30, 2016
-
Sjogren-Larsson syndrome (SLS) is a rare autosomal recessive neurocutaneous disorder with worldwide incidence of 0.4 per 100,000 people. It is characterized by the triad of congenital ichthyosis, spastic diplegia or quadriplegia, and mental retardation. Herein we report a 2-year-old male child with SLS, asthma, and recurrent pneumonia. SLS was confirmed by a molecular genetics study that revealed a deletion mutation...
- Original Article
- Neurology
- Clinical importance of F-waves as a prognostic factor in Guillain-Barré syndrome in children
- Eung-Bin Lee, Yun Young Lee, Jae Min Lee, Su Min Son, Su-Kyeong Hwang, Soonhak Kwon, Sae Yoon Kim
- Clin Exp Pediatr. 2016;59(6):271-275. Published online June 30, 2016
-
Purpose A limited number of studies have examined the link between F-wave abnormalities and clinical presentation in pediatric Guillain-Barré syndrome (GBS). Therefore, this study examined the importance of F-wave abnormalities as a prognostic factor in pediatric GBS patients.
Methods The records and electrodiagnostic studies (EDS) of 70 GBS patients were retrospectively evaluated, and divided into 2 groups according to the results of EDS....
- Review Article
- Nephrology (Genitourinary)
- Pathogenesis of minimal change nephrotic syndrome: an immunological concept
- Seong Heon Kim, Se Jin Park, Kyoung Hee Han, Andreas Kronbichler, Moin A. Saleem, Jun Oh, Beom Jin Lim, Jae Il Shin
- Clin Exp Pediatr. 2016;59(5):205-211. Published online May 31, 2016
-
Idiopathic nephrotic syndrome (INS) in children is characterized by massive proteinuria and hypoalbuminemia. Minimal change nephrotic syndrome (MCNS) is the most common form of INS in children. The pathogenesis of MCNS still remains unclear, however, several hypotheses have been recently proposed. For several decades, MCNS has been considered a T-cell disorder, which causes the impairment of the glomerular filtration barrier...
- Original Article
- Nephrology (Genitourinary)
- Acute tubular necrosis as a part of vancomycin induced drug rash with eosinophilia and systemic symptoms syndrome with coincident postinfectious glomerulonephritis
- Kyung Min Kim, Kyoung Sung, Hea Koung Yang, Seong Heon Kim, Hye Young Kim, Gil Ho Ban, Su Eun Park, Hyoung Doo Lee, Su Young Kim
- Clin Exp Pediatr. 2016;59(3):145-148. Published online March 31, 2016
-
Drug rash with eosinophilia and systemic symptoms (DRESS) syndrome is a rare and potentially fatal condition characterized by skin rash, fever, eosinophilia, and multiorgan involvement. Various drugs may be associated with this syndrome including carbamazepine, allopurinol, and sulfasalazine. Renal involvement in DRESS syndrome most commonly presents as acute kidney injury due to interstitial nephritis. An 11-year-old boy was referred to...
- Case Report
- Nephrology (Genitourinary)
- Recombinant Human Erythropoietin Therapy for a Jehovah's Witness Child With Severe Anemia due to Hemolytic-Uremic Syndrome
- Da Eun Woo, Jae Min Lee, Yu Kyung Kim, Yong Hoon Park
- Clin Exp Pediatr. 2016;59(2):100-103. Published online February 29, 2016
-
Patients with hemolytic-uremic syndrome (HUS) can rapidly develop profound anemia as the disease progresses, as a consequence of red blood cell (RBC) hemolysis and inadequate erythropoietin synthesis. Therefore, RBC transfusion should be considered in HUS patients with severe anemia to avoid cardiac or pulmonary complications. Most patients who are Jehovah's Witnesses refuse blood transfusion, even in the face of life-threatening...
- Original Article
- Cardiology
- Meta-analysis of factors predicting resistance to intravenous immunoglobulin treatment in patients with Kawasaki disease
- Jin-Young Baek, Min Seob Song
- Clin Exp Pediatr. 2016;59(2):80-90. Published online February 29, 2016
-
Purpose Studies have been conducted to identify predictive factors of resistance to intravenous immunoglobulin (IVIG) for Kawasaki disease (KD). However, the results are conflicting. This study aimed to identify laboratory factors predictive of resistance to high-dose IVIG for KD by performing meta-analysis of available studies using statistical techniques.
Methods All relevant scientific publications from 2006 to 2014 were identified through PubMed searches. For...
- Infantile Marfan syndrome in a Korean tertiary referral center
- Yeon Jeong Seo, Ko-Eun Lee, Gi Beom Kim, Bo Sang Kwon, Eun Jung Bae, Chung Il Noh
- Clin Exp Pediatr. 2016;59(2):59-64. Published online February 29, 2016
-
Purpose Infantile Marfan syndrome (MFS) is a rare congenital inheritable connective tissue disorder with poor prognosis. This study aimed to evaluate the cardiovascular manifestations and overall prognosis of infantile MFS diagnosed in a tertiary referral center in Korea.
Methods Eight patients diagnosed with infantile MFS between 2004 and 2014 were retrospectively evaluated.
Results Their median age at the time of diagnosis was 2.5 months (range,...
- Case Report
- Oncology
- Early onset of colorectal cancer in a 13-year-old girl with Lynch syndrome
- Do Hee Ahn, Jung Hee Rho, Hann Tchah, In-Sang Jeon
- Clin Exp Pediatr. 2016;59(1):40-42. Published online January 22, 2016
-
Lynch syndrome is the most common inherited colon cancer syndrome. Patients with Lynch syndrome develop a range of cancers including colorectal cancer (CRC) and carry a mutation on one of the mismatched repair (MMR) genes. Although CRC usually occurs after the fourth decade in patients with Lynch syndrome harboring a heterozygous MMR gene mutation, it can occur in children with...
-

-
-

-

-
Impact Factor4.2
-
6.52022CiteScore92nd percentilePowered by




")